

If you’re developing a health-related product, one of the most important questions you need to answer early is whether the FDA considers it a wellness device or a medical device. The distinction shapes everything — your regulatory pathway, your marketing claims, your labeling, and your liability exposure.
It’s also a question that’s easier to get wrong than most founders expect.
Getting the classification right from the start puts you in control of what comes next. Getting it wrong can mean rebranding, reworking your claims, or facing regulatory penalties after you’ve already gone to market. Here’s what you need to know.
What Is an FDA Wellness Device?
The FDA’s concept of a wellness device is grounded in a straightforward idea: that healthy lifestyle choices lead to better health outcomes. Wellness devices are designed to support those choices — not to diagnose, treat, or address a specific medical condition.
Common examples include fitness trackers, sleep monitors, food logging apps, and mental fitness games.
Key characteristics of an FDA wellness device:
- Not subject to FDA medical device regulations
- Designed to promote, track, or encourage healthy behaviors
- Poses low risk to the user or others
- Makes only general health and wellness claims
- Not intended to diagnose, treat, cure, or prevent a specific disease or condition
- May reference a disease or condition only in the context of reducing risk or supporting general management of that condition
That last point is where founders often run into trouble. The moment your product’s marketing or labeling crosses from general wellness language into clinical claims — even subtly — the FDA may view it as a medical device, regardless of how you’ve positioned it.
What Is a Medical Device?
Medical devices are formally defined in Section 201(h)(1) of the Food, Drug, and Cosmetic Act and are subject to extensive FDA regulation. Examples range from insulin pumps and CPAP machines to pulse oximeters and pacemakers.
A product is generally considered a medical device if it:
- Is intended to diagnose, treat, mitigate, cure, or prevent a specific disease or condition
- Carries meaningful risk to the user or others
- Is invasive or implantable
- Uses technology that poses risk, such as radiation
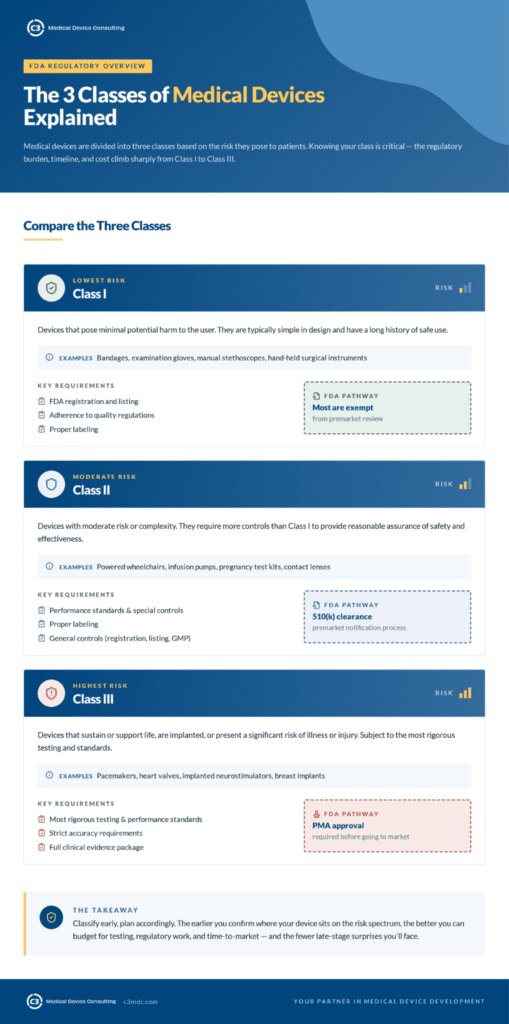
Medical devices are divided into three classes based on risk:
Class I — Lowest risk. Requires FDA registration and listing, adherence to quality regulations, and proper labeling. Most Class I devices are exempt from premarket review.
Class II — Moderate risk or complexity. Subject to performance standards and special controls, and requires proper labeling. Most Class II devices go through the 510(k) premarket notification process.
Class III — Highest risk. Includes devices that sustain life, are implanted, or present a significant risk of illness or injury. Requires FDA approval before going to market and is subject to the most rigorous testing, performance, and accuracy standards.
Understanding which class your device falls into is critical — because the regulatory burden, timeline, and cost increase significantly as you move from Class I to Class III.
Where the Line Gets Blurry
The FDA wellness device category exists in a gray zone that trips up a lot of early-stage teams. A product that looks like a wellness device on the surface can cross into medical device territory based entirely on how it’s marketed or what claims are made about it.
A few scenarios where the line commonly blurs:
Compression products. Graduated compression stockings marketed for general comfort are typically wellness products. The same product marketed specifically for diabetic patients or venous insufficiency may be classified as a medical device.
Software and apps. A general fitness app is usually a wellness product. An app that monitors a specific condition, provides clinical decision support, or affects the treatment of a disease may be regulated as a Software as a Medical Device (SaMD).
Wearables. A wearable that tracks general activity is a wellness device. One that monitors cardiac rhythm for the purpose of detecting arrhythmia is a medical device.
The through-line in all of these examples is intent and claims. The FDA looks at what you say your product does in your labeling, your marketing, your app descriptions, and even your website as much as it looks at what the product technically is.
How to Determine Your Device’s Classification
The FDA provides guidance for making this determination, and it’s worth working through carefully before you finalize your product concept or begin development. Key questions to answer:
- Does your product meet the FDA’s definition of a medical device under Section 201(h)(1) of the Food, Drug, and Cosmetic Act?
- Is there an existing product classification that applies to your device?
- If your product includes software, is that software considered a medical device or Software as a Medical Device?
- Is your product genuinely intended for general wellness, or does it make or imply specific clinical claims?
- Does your product contain drugs or pharmaceutical components?
If any of these questions leave you uncertain, that uncertainty is worth resolving before you go further. The cost of a misclassification compounds significantly as development progresses.
Why Getting This Right Early Matters
Beyond ethics and accuracy, there are real business consequences to misclassifying your device.
Patients and consumers make decisions based on how a product is classified, including whether it may be eligible for insurance coverage or reimbursement. Medical devices may qualify; wellness devices typically do not. If your target users expect reimbursement and your product isn’t classified to support that, it affects adoption.
From a legal and regulatory standpoint, marketing or selling a product that the FDA determines should have been classified as a medical device without the corresponding approval or clearance can result in warning letters, product recalls, injunctions, or civil penalties. These are not theoretical risks. They happen to real companies, including early-stage ones that simply didn’t know what they didn’t know.
Starting development with the right classification in place keeps your regulatory pathway clear, your claims accurate, and your business protected.
Not Sure Where Your Product Lands? C3 Can Help.
At C3, we work with founders and engineering teams at the earliest stages of development, including helping you think through FDA classification before you’ve committed to a design direction. If you’re not sure whether you’re building an FDA wellness device or a medical device, book a conversation with our team. Getting clarity now is far less costly than correcting course later.
You can also use our development cost estimator to get a sense of what a structured development program might look like once your classification is confirmed.
